Build practical regulatory strategies for digital health products, including medical devices, AI systems, IVDs, EHR systems, wellness applications, and all other healthcare-related software.
Regulatory strategy development
Product qualification and classification support
MDR and IVDR regulatory pathway support
AI Act assessment for low-risk, high-risk, and generative AI systems
EHDS considerations for EHR systems and wellness applications
CRA assessment for healthcare-related software
FDA and international regulatory strategy
Intended purpose and claims assessment
Gap assessments and regulatory readiness reviews
Technical documentation planning
Clinical, performance, and validation strategy support
Regulatory lifecycle and market expansion planning
Regulatory change and impact assessment support
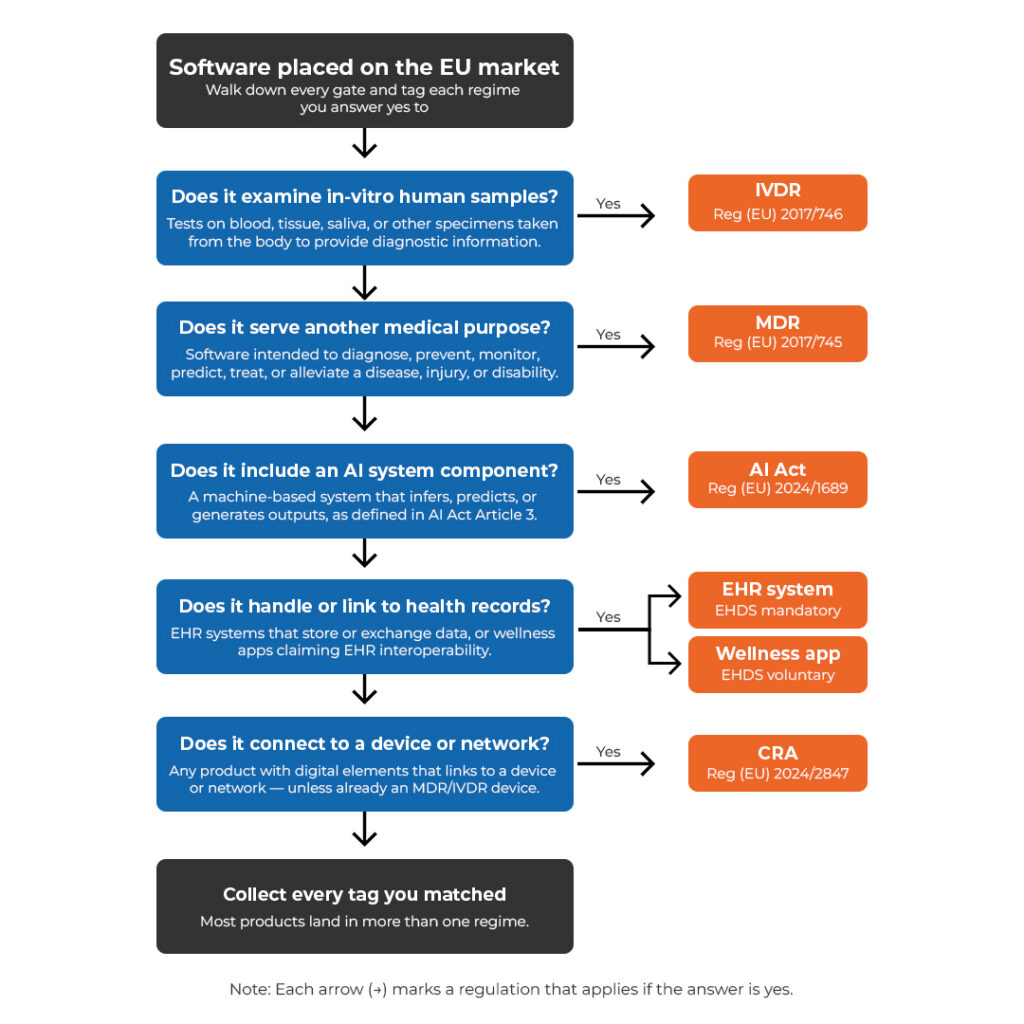
Digital health products rarely fall under a single regulatory framework.
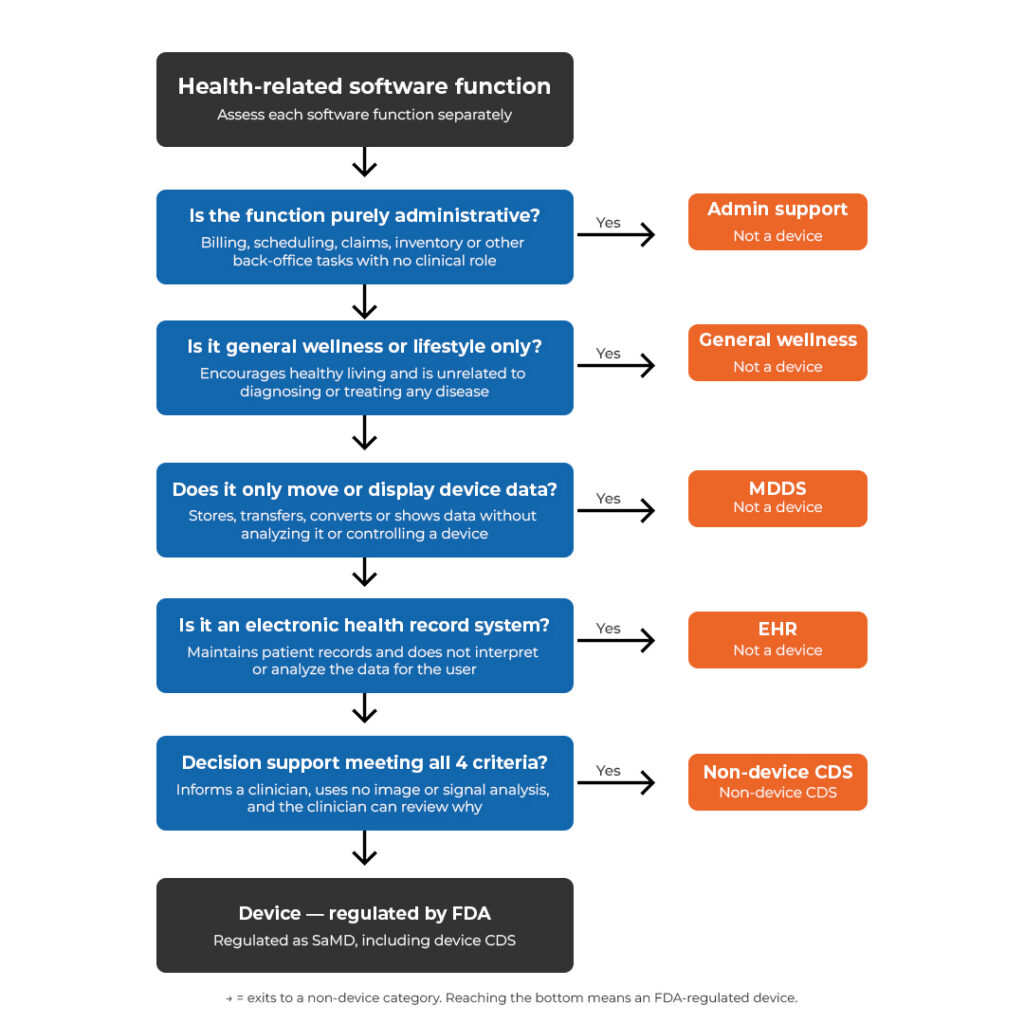
For example, in the European Union, a product may qualify as a SaMD under MDR, a high-risk AI system under the AI Act, and an EHR system under EHDS at the same time. Even if a product is a medical device, the EHDS applies too if the medical device processes electronic health records, and or connects to an EHR system. Other healthcare software, such as hospital maintenance planning tools, falls under the CRA instead. Getting this wrong early means rebuilding documentation, delaying market access, or discovering gaps during a Notified Body audit. In the United States, healthcare products may be regulated differently by the FDA based on their functionalities, for example, as clinical decision support software (either medical device or not), medical device database systems, or similar.
We support organisations in determining which frameworks apply to their product and what each one requires. This covers medical devices, in-vitro diagnostic medical devices, AI/ML-enabled medical devices, low and high-risk AI systems, including generative AI, EHR systems, wellness applications, SaMD, SaIVD, and other healthcare-related software.
From there, we build a regulatory strategy that covers qualification, classification, intended purpose definition, conformity assessment route, technical documentation planning, and validation strategy. We identify what regulations apply, which guidance documents, and applicable standards (e.g., ISO/IEC, EN, AAMI, ANSI, NEN, etc). Where multiple frameworks overlap, we map the requirements together so development and compliance work run in parallel rather than in sequence. The earlier this work starts, the more options remain open.
Most digital health products placed on the EU market fall under more than one framework. A single product can collect multiple tags.
FDA assesses each software function separately. A product with multiple functions may land in more than one outcome category.


Explore our blog posts on MDR, IVDR, and AI Act compliance to stay ahead of regulatory changes.
Find answers to common questions about our services, compliance processes, and how we can assist your business.
The intended purpose and indications for use you document at the start determine which regulations apply, how the product is classified, and what evidence you need to reach the market. Changing these later means reworking documentation, retesting, and in some cases redesigning the product.
Starting with a clear regulatory strategy keeps development aligned with what regulators will expect.
It depends on the intended purpose, claims, and target users. SaMD and SaIVD fall under MDR and IVDR, respectively. AI/ML-enabled medical devices and high-risk AI systems are subject to both MDR and IVDR and the AI Act. EHR systems and wellness applications fall under EHDS, however, also medical devices processing electronic health records are subject to compliance with EHDS. Other healthcare-related software, such as hospital maintenance planning tools, may be subject to the CRA. Many products are subject to more than one framework simultaneously.
They address different aspects of the same product. MDR and IVDR govern safety and performance as a medical device or IVD. The AI Act adds requirements around data governance, transparency, and human oversight for AI systems. EHDS covers interoperability and data access for EHR systems and wellness applications, and lays down additional requirements for medical devices around processing of electronic health records. CRA sets cybersecurity requirements for connected products not covered by MDR or IVDR.
Where frameworks overlap, requirements must be addressed together, not sequentially.
Yes. Guidance documents, harmonised standards, common specifications, and regulatory interpretations continue to evolve. The AI Act, EHDS, and CRA are still being implemented, with delegated acts and technical specifications expected over the coming years. Regulatory strategy is an ongoing process, not a document produced once at the start.
(Existing FAQ from MedQAIR FAQ page, relevant to this service page)
The European Union is today our home market, where we maintain close contact with Notified Bodies, and especially those that are experienced in the field of SaMD’s and SaIVDs.
However, we do have significant expertise in other regulatory jurisdictions too, especially in the field of software as a medical device (SaMD, SaIVD). We have supported multiple types of FDA submissions throughout our individual careers, such as Pre-submissions, 510(k)’s, 510(k)’s subject to special controls, such as reader-studies (MRMC), De Novo applications, PMA submissions, and 513(g)’s.
We have also performed applications in other regulatory jurisdictions and supported the implementation of MDSAP-compliant management systems.
(Existing FAQ from MedQAIR FAQ page, relevant to this service page)
As a first step, it is important to document what your product will be used for carefully (its ‘intended purpose’ or ‘intended use’ and ‘indications for use’), and what claims will be associated with the product. Based on that information, it is highly recommended to document a regulatory strategy, assess whether local legislation may apply to the product, and determine how the product is classified under such legislation (‘product qualification and classification’).
The landscape of digital legislative frameworks is becoming increasingly complex, and often more than one legislative instrument may apply. For example, in the European Union, a product may qualify as a medical device (SaMD, SaIVD, SAIeMD), and or artificial intelligence system (potentially being High-Risk), and be subjected to medical device legislation (MDR 2017/745 or IVDR 2017/746), artificial intelligence legislation (AI Act 2024/1689) and healthcare interoperability legislation (EHDS 2025/327).
The applicable legislation may be supported by various standards and guidance documents, which set out further requirements applicable to the product. Having a thorough understanding of all these aspects is important prior to the start of the development of a product, to ensure that by the end of the development process, all regulatory requirements have been met.
An app is a medical device under EU MDR if it has a medical purpose as defined in Article 2(1), such as diagnosis, monitoring, prediction, prognosis, treatment, or alleviation of disease. Pure wellness, fitness, or lifestyle apps are generally not medical devices. The MDCG 2019-11 guidance provides decision trees for borderline software. If your app processes patient data to inform a clinical decision, it is almost certainly software as a medical device (SaMD) and will require CE marking under MDR.
Software as a Medical Device (SaMD) is software that performs a medical function on its own, without being part of a hardware medical device, for example, a smartphone app that analyzes ECG data. Software in a Medical Device (SiMD) is software that is embedded in or controls a physical medical device, such as the firmware of an insulin pump. Both must comply with IEC 62304, but the risk classification, clinical evaluation pathway, and cybersecurity expectations may differ.
The total cost for CE marking under MDR depends heavily on device class and software complexity. For a Class IIa SaMD, expect roughly €80,000–€250,000 for the full first-cycle effort, covering QMS implementation, technical documentation, clinical evaluation, Notified Body fees, and consulting. Class IIb and III devices, and devices requiring clinical investigation, can exceed €500,000. Notified Body fees alone typically range from €30,000–€80,000 for initial certification, with annual surveillance audits thereafter.
Product qualification determines whether a product falls within the scope of a regulation at all. Classification then determines the risk category within that regulation. A SaMD that qualifies under MDR must still be classified as Class I, IIa, IIb, or III based on its intended purpose and risk profile. Getting the qualification wrong means the classification work that follows is built on an incorrect foundation.
Intended purpose documentation informs classification, which in turn determines the conformity assessment route. Under MDR, a Class IIa SaMD follows a different pathway to a Class IIb or III device. Under the AI Act, how the intended purpose is framed affects whether a system is treated as high-risk under Annex III. Small differences in how claims are written can materially change the evidence burden and timelines.
MedQAIR builds multi-market regulatory strategies by mapping requirements across jurisdictions before development starts, identifying where EU and FDA documentation can be aligned and where genuine differences require separate approaches. For AI/ML-enabled medical devices, this includes mapping MDR and AI Act requirements against FDA classification and predicate strategy in parallel, so that development decisions serve both submissions rather than creating conflicts that surface late.
Cookies help us improve your experience on our website. By using our site, you consent to the use of cookies as described in this policy.






