
Unique Device Identification is being implemented on a global scale at a fast pace to allow authorities and economic operators to keep track of all the medical devices used in healthcare. What is Unique Device Identification, and what should you consider for medical devices?
Imagine a method for detailed tracing for every medical device—from surgical tools to diagnostic tests. Thanks to the EU’s MDR and IVDR regulations, each device gets a unique code, making it easy to trace its journey from the factory to the patient. This system boosts safety, transparency, and trust in medical care. Let’s dive into how UDI is revolutionising the way we see and use medical devices!
What is the Unique Device Identification (UDI) System under the EU MDR and IVDR?
The Unique Device Identification (UDI) System under the EU Medical Devices Regulations 2017/745 (MDR) and 2017/746 (IVDR) is a system designed to enhance the identification and traceability of medical devices.
The UDI is a series of numeric or alphanumeric characters generated according to a globally accepted device identification and coding standard. It enables the clear and precise identification of a specific medical device on the market.
The UDI system encompasses all devices, except for custom-made devices and those used for performance studies or investigations.
Why is UDI important?
The UDI System is a way to give every medical device a unique identifier. Think of it like a barcode or string of numbers on products you buy in a store, but more detailed and standardized for medical devices.
Benefits of UDI
UDI is important because of the following benefits:
- Patient Safety: Helps in post-market safety. Authorities can quickly identify faulty devices and take them off the market. It reduces medical errors and combat the use of counterfeit devices.
- Traceability: Makes it easier to track devices throughout their lifecycle—from production to end-use.
- Regulation Compliance: Ensures that all medical devices meet EU standards for safety and performance.
- Better procurement and waste disposal: Optimize procurement, waste disposal procedures, and inventory management for healthcare institutions and various economic operators.
The Essential Components of the UDI System
Unique Identifier: Each medical device gets a unique code that consists of:
Basic UDI-DI serves as the primary identifier for a group or family of products that share the same intended purpose, risk class, and essential design and manufacturing characteristics. The Basic UDI-DI does not appear on the product packaging. It is an invisible number for your customers and is used solely for administrative purposes, e.g. in the technical documentation, EUDAMED and the Declaration of Conformity and the CE certificate (as applicable).
UDI-DI (Device Identifier) is specific to each device and or packaging configuration and contains information about the manufacturer, authorized representative, device model, and other relevant details. The DI part of the UDI number remains static across all units of the same device model, ensuring that it does not change for each device manufactured, in line with the same Technical Documentation
For stand-alone software medical devices, the UDI-DI might be considered the Minor version of a Major.Minor.Patch configuration.
UDI-PI (Production Identifier) captures the variable details of a device’s production, including manufacturing date, expiration date, lot number, and serial number. Unlike the static Device Identifier, the PI varies with each device, reflecting the specific production conditions for that unit. One UDI-DI can have numerous variants of UDI-PI.
For stand-alone software medical devices, the UDI-PI might be considered the Patch version in a Major.Minor.Patch configuration.
UDI Carrier
A UDI carrier is how the UDI code is presented, combining an automatically readable part like a barcode (for most devices, e.g. it may not be applied for stand-alone software) with a human-readable part such as numeric codes for easy identification and interpretation. It serves as the vehicle through which the device’s unique identification data is conveyed and accessed for regulatory, tracking, and informational purposes.
The Device Identifier (DI) and Production Identifier (PI) together form the complete UDI, with the DI appearing first followed by the PI.

You might have noticed two kinds of presentations of UDI in the above image.
They are:
-
Automatic Identification and Data Capture (AIDC)
AIDC refers to technologies used to automatically identify objects, collect data about them, and enter that data directly into computer systems without human involvement. One or two-dimensional barcodes can be rapidly scanned using a barcode reader, seamlessly integrating into electronic patient records or digital systems for straightforward device identification.
Other AIDC technologies include barcodes, Radio Frequency Identification (RFID), biometrics, magnetic strips, smart cards, Optical Character Recognition (OCR), etc.
-
Human Readable Interpretation (HRI)
Human Readable Interpretation (HRI) refers to the representation of data in a format that is easily readable and understandable by humans. For example, in a barcode label, the HRI is the text or numbers printed below or alongside the barcode symbol. This human-readable information provides essential details that can be quickly interpreted without the need for a barcode scanner or other specialized equipment.
UDI Placement Requirements
Your UDI should be positioned on the product labels and packaging. For reusable devices, it should also be directly engraved on the device itself, typically via laser etching, as the packaging is discarded after initial use.
If there isn’t enough space on the primary packaging, as is often the case for many small IVDs, the UDI can be placed on the next packaging level. When only one UDI format can fit, Automatic Identification and Data Capture (AIDC) takes priority, except for devices intended for use outside healthcare facilities, where Human Readable Interpretation (HRI) format takes precedence.
For software medical devices, the UDI-DI and UDI-PI (jointly ‘UDI’) could be positioned on the product label, e.g. on an about page, or reports generated by the software. When it is embedded in the generated results for software, it will easily trace back the results to the version of the software used to generate it.
EUDAMED Entry
Information about the device and its identifier is entered into a central EUDAMED (European Database on Medical Devices) database, as per Article 28 of the MDR and Article 25 of the IVDR. This database is accessible to regulators, healthcare providers, and the public.
EUDAMED is a secure web platform designed for capturing and sharing data about medical devices available on the EU market, as well as those undergoing clinical investigations.
The manufacturer bears responsibility for meeting all UDI-related requirements outlined above before placing the products on the market.
UDI - Mandatory Deadlines
The mandatory deadline for a device to comply with the UDI (Unique Device Identification) requirements varies depending on the device type:
- The obligation to assign UDI applies starting from the date of application of the two new Regulations: 26 May 2021 for medical devices and 26 May 2022 for in vitro diagnostic medical devices.
The requirement to submit UDI data to the EUDAMED database starts on 26 November 2022 for medical devices and 26 November 2023 for in vitro diagnostic medical devices, contingent upon EUDAMED being fully operational before the Regulation’s effective date. If EUDAMED is not fully functional by then, this obligation applies 24 months after EUDAMED achieves full functionality.
Manufacturers may voluntarily comply with registration obligations starting from 26 May 2021 for medical devices and 26 May 2022 for in vitro diagnostic medical devices. Full registration of devices (Article 29 of MDR and Article 26 of IVDR) is required before any relevant serious incident can be registered in Eudamed, provided Eudamed is fully functional after these dates.
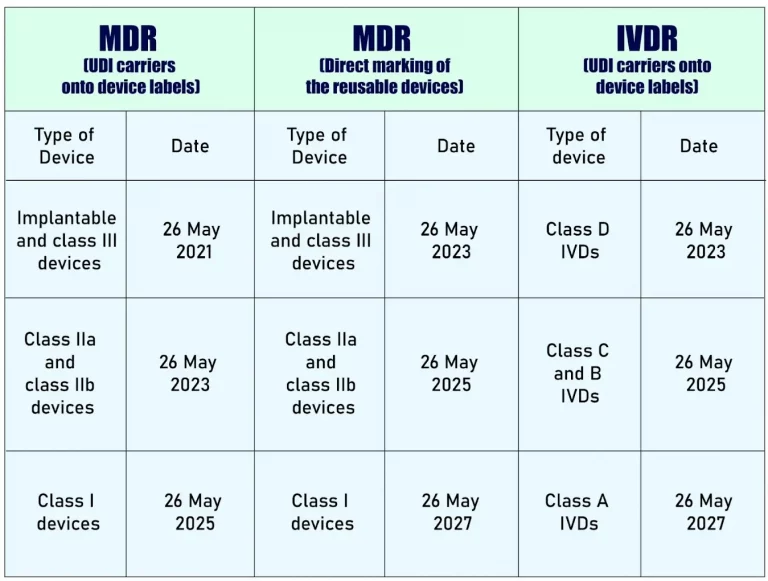
- Placing UDI-carriers on the labels of devices (2017/745 (‘MDR’): 26 May 2021 for Implantable devices and Class III devices; 26 May 2023 for Class IIa and Class IIb devices; and 26 May 2025 for Class I devices. Direct marking of the reusable devices (MDR Article 123(3)(g), Article 27(4)) for the same categories respectively are 26 May 2023; 26 May 2025; and 26 May 2027.
- Placing UDI-carriers on the labels of devices (2017/746 (IVDR): 26 May 2023 for Class D IVDs, 26 May 2025 for Class C and B IVDs; and 26 May 2027 for Class A IVDs.
Ref – https://webgate.ec.europa.eu/udi-helpdesk/en/udi-carrier/deadlines.html
The timelines for placing UDI Carriers
How Does the UDI System Work?
Assignment of UDIs by the issuing entities
The UDI issuing authorities as designated by the Commission (under MDR Article 27(2)/IVDR Article 24(2)) are:
- GS1 AISBL
- Health Industry Business Communications Council (HIBCC)
- International Council for Commonality in Blood Banking Automation (ICCBBA)
- Informationsstelle für Arzneispezialitäten (IFA) GmbH
Entities operating UDI assignment systems must ensure uniqueness. These issuing organizations must maintain this role for at least 10 years.
TIP: it may be worthwhile to compare these organizations and their offerings, prices, and contractual conditions differ significantly.
Adding UDIs to devices and packaging by manufacturers
Manufacturers request a unique identification number from allocation offices for each medical device and higher-level packaging (excluding shipping containers). The MDR exempts UDIs only for custom-made devices and clinical trial products.
Manufacturers are required to incorporate the Basic UDI-DI into their declaration of conformity and maintain a comprehensive list of all assigned UDIs in the technical documentation for each medical device.
Storing of the UDIs by economic operators
All economic operators and health institutions are required to store and maintain the UDI of class III implantable devices they supply or receive, preferably through electronic means.
Operating UDI database by the EU
The EU is required to maintain a database that stores all UDIs. This database outlines the essential attributes that must be recorded for each medical device. Access to this database is public and free of charge.
The Global UDI Landscape
The field of Unique Device Identification (UDI) is continually evolving and varies in its level of advancement across different countries.
United States
- Regulation: The U.S. Food and Drug Administration (FDA) finalized its UDI rule in September 2013.
- Implementation: Compliance has been phased based on the device’s risk class:
- Class III devices: Since September 24, 2014
- Class II devices: Since September 24, 2016
- Class I and unclassified devices: Since September 24, 2020
- Database: The Global Unique Device Identification Database (GUDID) stores UDI information.
- Application: UDIs (in both human- and machine-readable formats) must appear on device labels, packaging, and sometimes directly on devices.
Switzerland
- Regulation: Switzerland aligns with the European Union’s UDI regulations under the EU Medical Device Regulation (MDR) and In Vitro Diagnostic Regulation (IVDR).
- Implementation: The MDR came into effect on May 26, 2021, and the IVDR on May 26, 2022, with transition periods for certain devices.
- Database: In April 2022, Swissmedic began designing the Swissdamed database, a comprehensive Swiss resource for medical devices and in vitro diagnostic devices. This database will offer detailed insights into the life cycle of these devices in Switzerland. Swissdamed’s rollout will occur in phases, with the latest update in June 2024, where Swiss manufacturers tested the mass upload of regulatory devices using the XML format.
China
- Regulation: The National Medical Products Administration (NMPA) oversees UDI implementation.
- Implementation: China has a phased approach:
- High-risk Class III devices: Since January 1, 2021
- Class II devices: June 1, 2024
- Other devices to follow based on NMPA’s schedule.
- Database: The China Unique Device Identification Database (CUDID) is used for data submission.
- Application: UDIs must be on device labels and packaging, with data submitted to CUDID.
Now, What?
Are you a producer of medical devices or in vitro diagnostics facing challenges with MDR and IVDR compliance? Are you finding it difficult to implement the UDI system for your products?
MedQAIR can assist you. Our regulatory experts are here to help you navigate and fulfill all UDI-related requirements and deadlines.
Contact us today to see how we can support your business!


