
On January 7, 2025, the MDCG 2023-3, a guidance document from the Medical Device Coordination Group (MDCG) of the European Union, was updated to Revision 2. It provides key updates to the vigilance management of medical devices (MDs) and in vitro diagnostic devices (IVDs).
This document, available here, provides questions and answers related to vigilance terms and concepts found in Section 2 of Chapter VII of the Medical Device Regulation (MDR) 2017/745 and In Vitro Diagnostic Medical Devices Regulation (IVDR) 2017/746.
It offers comprehensive guidance on the application of vigilance requirements, emphasizing the definitions, reporting criteria, and procedures associated with incidents and serious incidents for medical devices and in vitro diagnostic devices. It also streamlines reporting processes through Eudamed and provides enhanced technical references.
The document is intended for competent authorities, economic operators, and other relevant stakeholders, providing essential guidance on the implementation of vigilance requirements under the MDR and IVDR regulations.
Key Takeaways from the MDCG 2023-3 Rev.2 Guidance
The latest revision pertains mainly to the following areas:
Question 1, Footnote 8
Footnote 8 has been updated to reflect the latest amendments under Regulation (EU) 2024/1860, ensuring consistency with the gradual roll-out of Eudamed, new obligations for supply continuity notifications, and transitional provisions for specific IVDs.
Question 21
The reference to the “Eudamed Vigilance (VGL) module” has been revised to “Eudamed Post-Market Surveillance and Vigilance module (VGL module)”, ensuring clarity and alignment with the latest regulatory framework.
Question 21 now provides clearer guidance on utilizing Eudamed for reporting incidents and serious incidents, promoting a standardized and efficient vigilance process across the EU. This ensures greater transparency, streamlined reporting, and improved regulatory compliance for medical device manufacturers.
Footnote 34
The guide now specifies that manufacturers must “allow 48 hours (equivalent to two weekdays)” for reporting incidents, replacing the previous reference to “48 working hours” for greater clarity.
New precise references offer enhanced regulatory guidance and best practices for compliance and data management, ensuring manufacturers have a clearer direction on vigilance requirements.
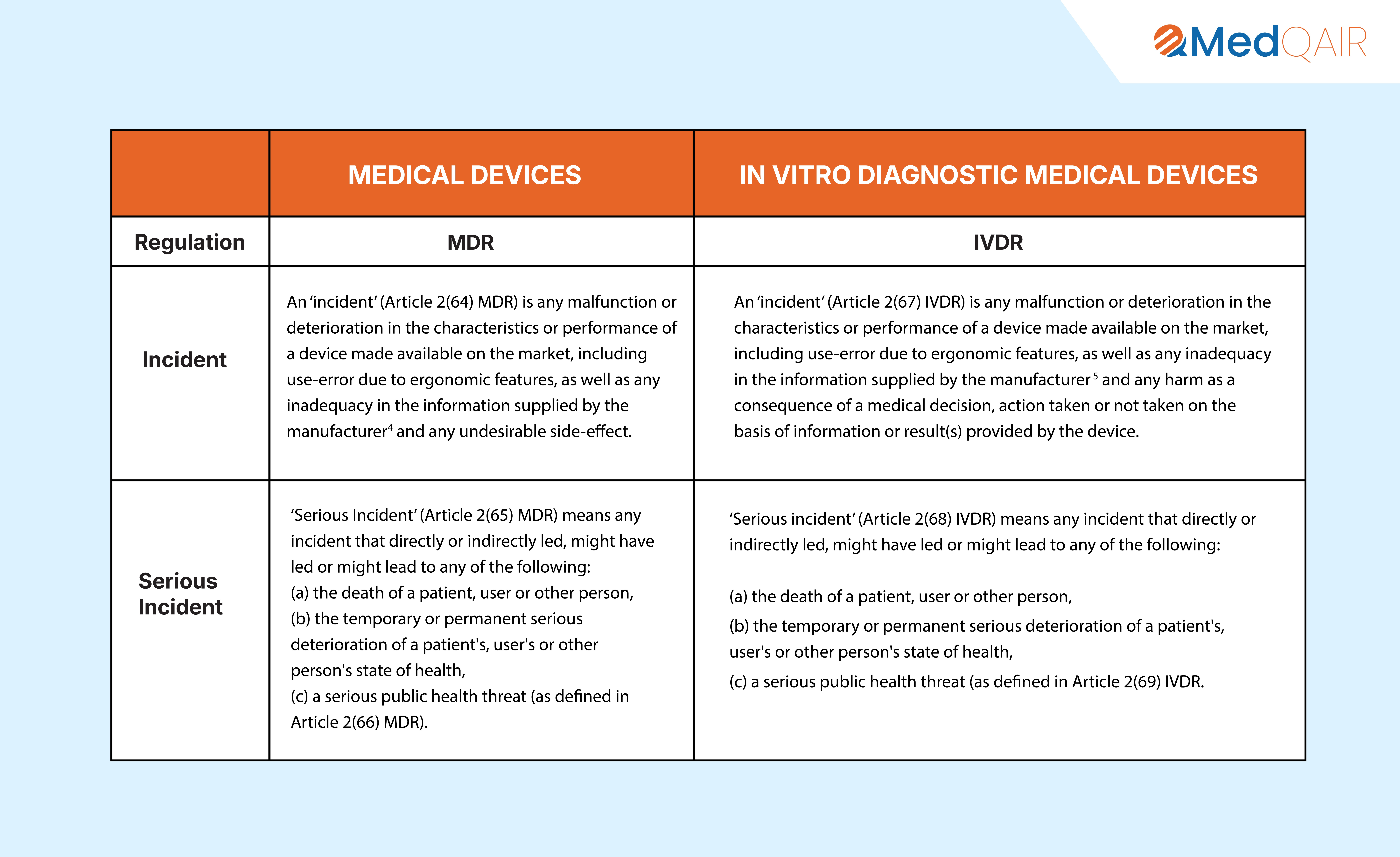
Refined Definitions of "Incident" and "Serious Incident"
Question 1 now also includes minor but impactful modifications to the definitions table, improving the readability and interpretation of key terms. The latest update enhances clarity and differentiation between “Incident” and “Serious Incident”, ensuring precise reporting and compliance. A detailed comparison table remains a key feature, providing structured guidance for manufacturers on assessing and categorizing events accurately.
If you need assistance understanding how these updates could impact your medical device applications, reach out to us for expert support and tailored advice.


