
Navigating post-market compliance under MDR/IVDR has never been more complex. As regulatory demands rise, responsibility no longer rests solely with the manufacturer, it extends across the entire distribution chain. Roles such as the AR and importers are now central to compliance and liability. In this landscape, the Medical Device Information System (MDIS) for medical devices play a pivotal role in reinforcing traceability and transparency across medical device distribution in EU markets.
In this post, we’ll explore how authorised representatives and importers operate under current regulations, the challenges they face in managing post-market obligations, and how MDIS offers a structured, scalable solution.
1. The Evolving Role of Authorised Representatives and Importers
Under the EU MDR (Article 11) and IVDR, the EU MDR authorised representative is legally mandated to act on behalf of non-EU manufacturers. This role includes reviewing technical documentation, ensuring registration in EUDAMED, and acting as a liaison for authorities. ARs also share liability for defective devices if they fail compliance obligations.
Similarly, importers under Article 13 (and distributors per Article 14) must verify CE marking, UDI presence, technical documentation availability, and correct labelling. They must maintain records, cooperate with authorities, and act on suspected non-compliance.
These roles are no longer optional add-ons, they are critical economic operators in the regulatory framework, fully integrated into post-market surveillance workflows. Yet, many still rely on fragmented tools and disconnected systems.
2. The Challenges of Current Post-Market Practices
Disconnected Documentation and Verification
Across the life cycle of a device, economic operators (manufacturer, AR, importer, distributor) need permanent access to the full or parts of technical documentation, IFUs, labels, and UDI records. Many organisations still use email, Excel, or siloed folders – creating delays, errors, and compliance gaps.
Duplication of Data and Effort
Each UDI-DI registration or product update must be reflected in regulatory databases such as EUDAMED, GUDID, or other national databases. Without a centralized system, the ARs and importers repeatedly request documents, causing duplicate effort and fragmented workflows.
Liability Exposure for Authorised Representatives
Without timely access to manufacturer documentation, EU MDR authorised representatives may be unable to verify updates or registrations. In case of audit or incident, this lack of transparency can mean shared liability.
Inefficient Incident Response
In post-market incidents, delays in sharing the latest documentation or UDI history across operators can slow vigilance reporting, hamper root cause analysis, or risk regulatory penalties.
3. What is MDIS and Why It Matters
MDIS (Medical Device Information System) is a purpose-built platform developed by MedQAIR to address these inefficiencies directly. It is designed specifically for all economic operators within the medical device distribution in the EU ecosystem.
From launch, MDIS focused on streamlining:
- UDI-DI tracking and management
- Secured document sharing (technical files, IFUs, labels)
- Verified workflows between economic operators
- Automated EUDAMED registration and updates
- Audit-ready archives and revision control
Leon Doorn, MedQAIR’s CEO, highlights that MDIS “streamlines the management of UDI-DIs and technical documentation during post-market stages” for all stakeholders.
4. How MDIS Supports the Medical Device Authorised Representative
MDIS provides a centralized, auditable environment tailored to the needs of the medical device authorised representative:
Streamlined Documentation Access
Medical Device Authorised Representatives (ARs) often face delays waiting for essential documentation like IFUs and labels. With MDIS for medical devices, they gain real-time, role-based access to all relevant files, cutting down manual follow-ups and ensuring faster time to compliance.
Built-in Verification and Review Workflows
Compliance under MDR Article 11 requires structured oversight. MDIS enables ARs to efficiently review, approve, and sign off documentation, CE certificates, and EUDAMED submissions through systemised workflows, helping maintain strict conformity with MDR requirements.
Reducing Liability Through Digital Proof
ARs operate under significant legal liability in the EU. MDIS helps mitigate this risk by offering timestamped audit trails, approval records, and version control. This gives ARs the digital proof they need to demonstrate due diligence and traceability during inspections or audits.
Collaboration Without Complexity
Managing multiple manufacturers and importers can be a logistical nightmare. MDIS simplifies collaboration by allowing ARs to invite stakeholders directly into the platform, keeping everyone aligned on documentation, responsibilities, and compliance timelines without messy email threads or file exchanges.
5. Empowering Importers with MDIS
Importers, especially those representing multiple manufacturers, face a challenge in tracking medical device documentation flows across jurisdictions. MDIS helps by:
- Consolidating manufacturer data in one dashboard
- Assigning verification tasks aligned with Articles 13 and 14
- Storing labels and IFU translations relevant to local markets
- Ensuring importers can trace incoming products and regulatory statuses efficiently
Through MDIS, importers can verify UDI compliance, label conformity, storage conditions, and regulatory registration – all with traceable logs and shared visibility.
6. Streamlining Post-Market Coordination Across Operators
One of the core strengths of MDIS is unifying economic operators:
- Digital Document Exchange: MDIS eliminates email chains or file servers, allowing authorised representatives, importers, and distributors to access role-specific information in real time.
- Automated Database Integration: UDI registration and EUDAMED uploads are automated, reducing manual errors and ensuring regulatory consistency.
- Incident Management and Vigilance: In case of adverse events, MDIS ensures all stakeholders access the latest documentation and UDI history promptly, facilitating IMDRF-coded reporting and compliance with submission timelines.
Leon Doorn emphasizes that MDIS connects all economic operators to maintain alignment and compliance throughout the post-market phase.
7. Real-World Scenarios: MDIS in Action
Scenario 1: Multi-Manufacturer AR Oversight
An AR represents three non-EU manufacturers supplying different device types. Traditionally, documentation arrives by email and lacks consistent updates. With MDIS, all documentation is centrally uploaded by each manufacturer; the AR validates, approves, and tracks EUDAMED registration in one platform.
Scenario 2: Importer Managing Labeling Updates
An importer receives devices from various sources. When IFU updates or UDI-DI changes occur, MDIS notifies them and stores version-controlled labels – ensuring they only distribute compliant documents.
Scenario 3: Post-Market Vigilance Coordination
A field safety incident triggers a vigilance report. Through MDIS, the importer, AR, and manufacturer collaborate instantly, with shared access to event documentation and UDI history – leading to faster submission to EUDAMED and reduced regulatory fallout.
8. Why MDIS Outperforms Traditional Systems
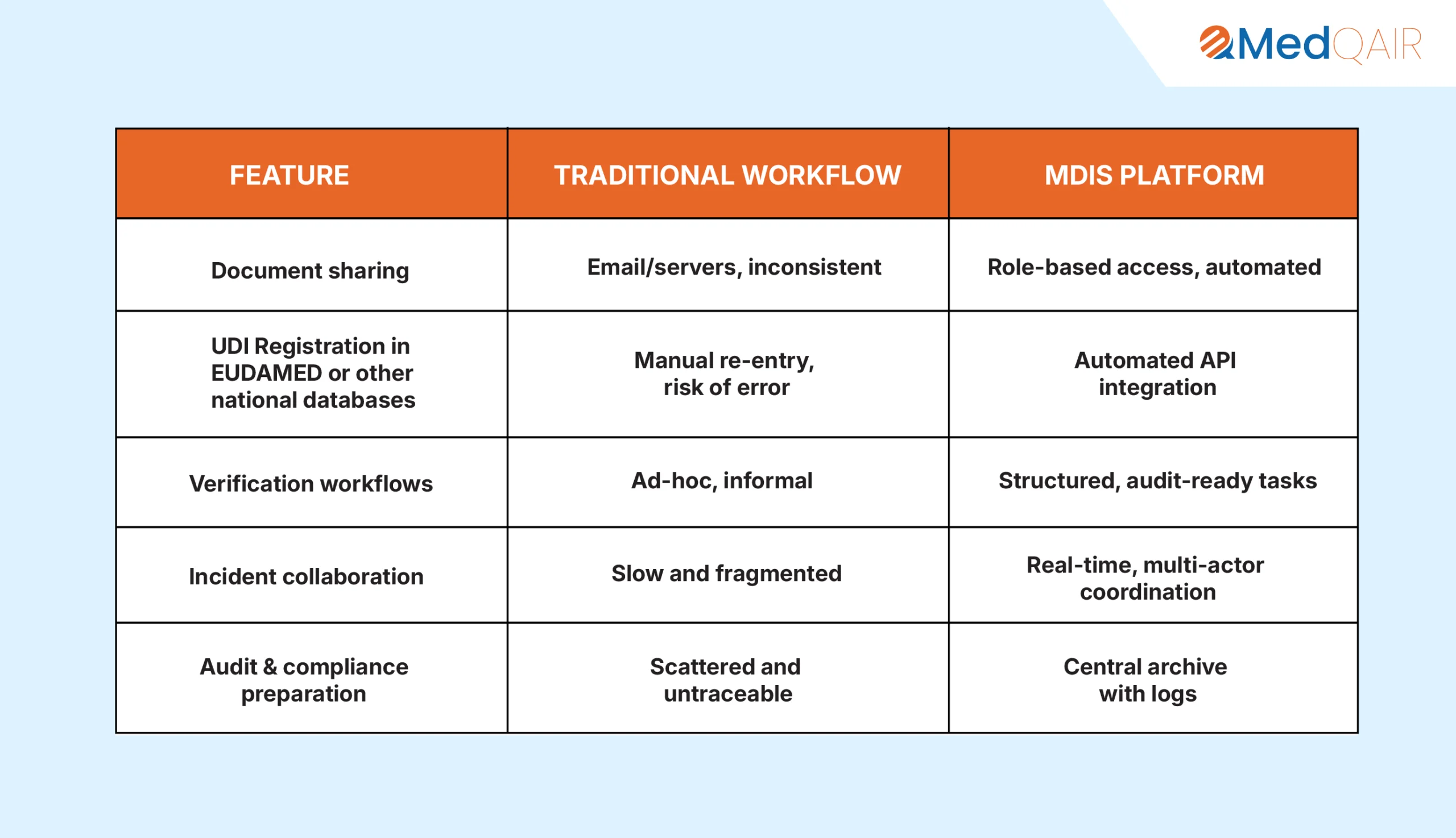
9. Final Thoughts: Combining Human Expertise with Digital Precision
The rise in regulatory complexity demands both human insight and digital continuity. As Leon Doorn writes, compliance is now a collective responsibility – but when operators work in isolation, gaps appear.
By combining MedQAIR’s regulatory expertise with MDIS’s systematic architecture, organisations can move from reactive, fragmented processes to proactive, traceable compliance. For medical device authorised representatives and importers committed to robust post-market vigilance, MDIS offers not just a tool – but a collaborative regulatory ecosystem.
Ready to Transform Your Post-Market Compliance?
Reach out to MedQAIR to:
- Discover how MDIS for medical devices secures operator coordination
- Learn how it supports medical device distribution in EU contexts
- Schedule a free demo and explore role-specific workflows for ARs and importers


