Within the legislation, the definition of a medical device Distributor is explained as per the below.

There are a few takeaways from the definition that is provided:
- Distributors are parties that make devices available on the European Union market up until the point of putting the devices into service, and
- A Manufacturer or Importer does not take on the role of a ‘Distributor’
In simpler terms, Distributors are entities that store and supply medical devices to retailers, healthcare providers, or directly to end-users. In several European Union countries, Distributors are required to register themselves with the local authorities. If the Distributor receives the product from a non-EU region, they also may take on the role of importer for the products.
As a Manufacturer using a distribution partner (or multiple), it may be worthwhile to verify whether Distributors have actually complied with their local registration requirements as part of the selection process and specifically in the event of software provided over online distribution platforms, where this may not be straightforward.
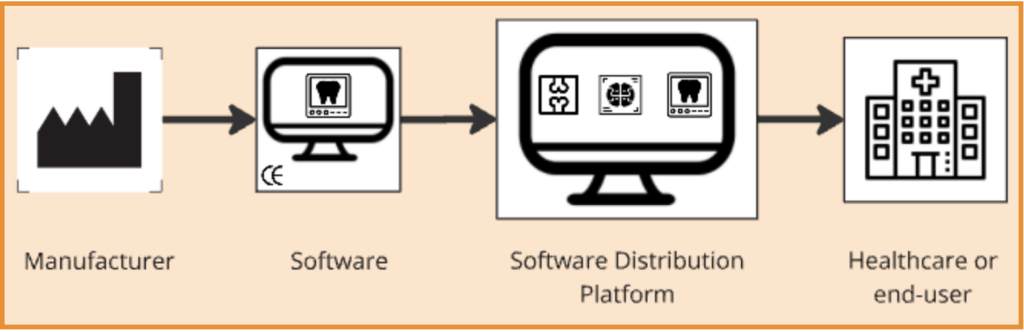
For software medical devices, there are more interesting notes. For example, if and where medical device software is integrated into a larger system provided to the market by a third party, the provider of the entire system (e.g. a PACS with embedded third-party AI tools) may be considered the ‘Distributor’. Similarly, when making medical device software available through a platform (e.g. the Apple App Store or Google Play Store) these may also take on the role of medical device Distributor.
A more detailed analysis of the requirements incumbent on those parties (specifically Apple and Google) has been investigated in detail by Sadare et al, in 2023. A quick review of the Apple App Store however quickly reveals that for medical device apps critical information is missing, e.g. the relevant symbols as mandated under the MDR are not displayed in the App Store, as for the CE mark of the products.
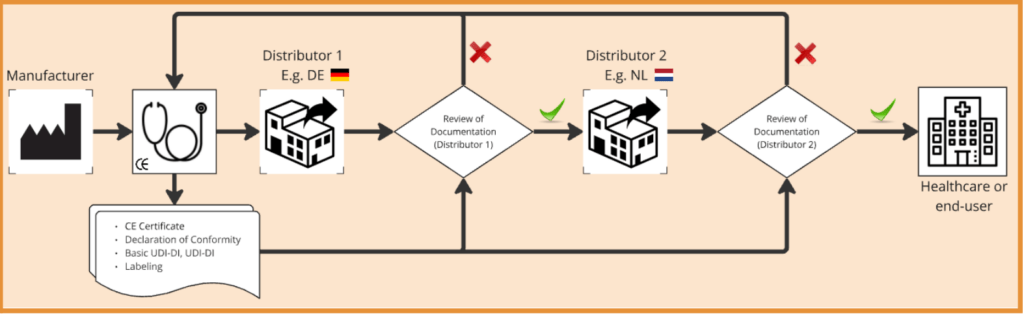
When a Distributor starts their distribution activities for a medical device Manufacturer they must ensure that the CE marking and EU conformity declaration are present and compliant. This may not only apply to the first-time distribution only, but also for any consequent updates and releases of new CE certificates and Declarations of Conformity. In the event of software with frequent updates, this process may require fast interactions between both parties.
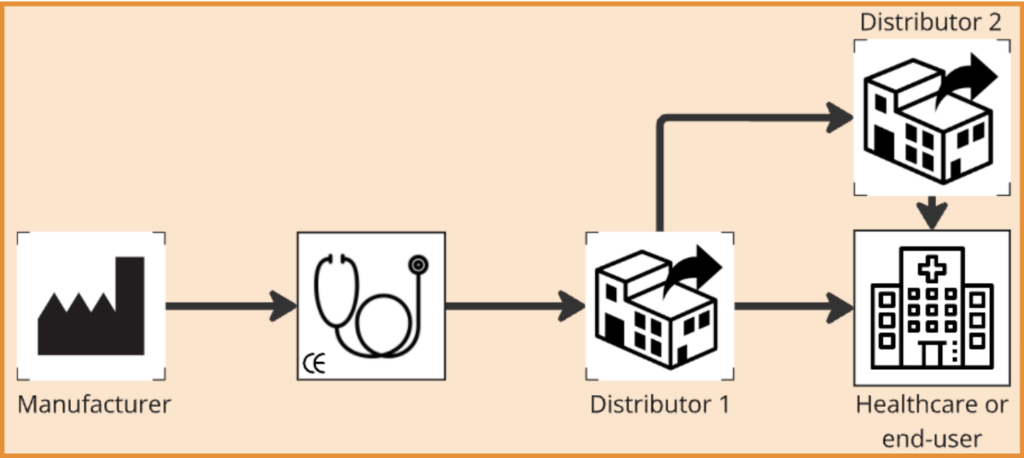
Consequently, the Distributor is tasked to ensure that the labeling is adequate, and for the countries to which they distribute, taking language requirements into consideration may need to be considered. If there are multiple Distributors along the full distribution chain, it may require different verification steps as each Distributor bears the same responsibilities. Where an importer is involved, the Distributor should also take note of whether their details are clarified.
As applicable, sampling procedures may be applied at a Distributor.
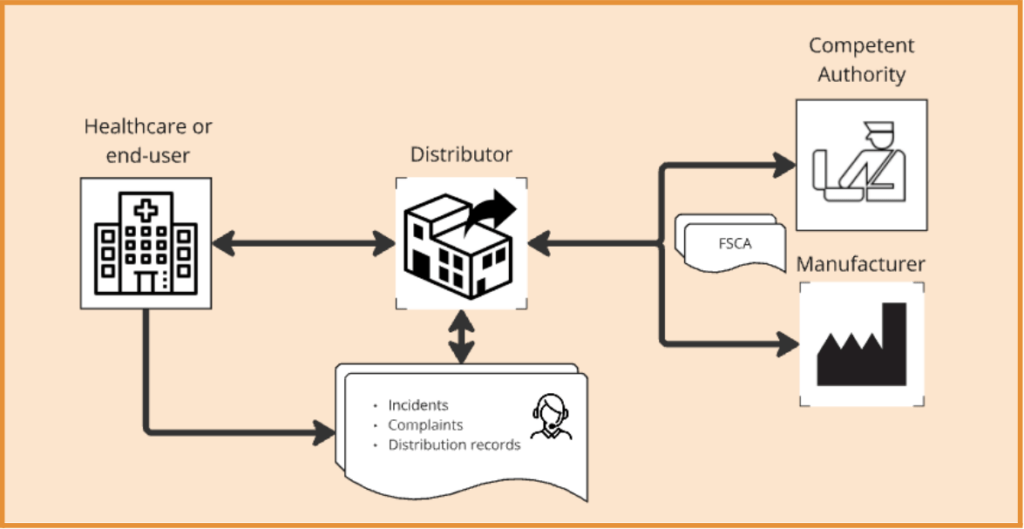
Once the device has been made available to the end-user, the Distributor continues to play a role in the distribution process. For example, they should ensure that they can respond to questions from the market, such as complaints and other forms of feedback provided by the end-user. Complaints and potential reported incidents will need to be logged by the Distributor. At the same time, they have the responsibility to support the Manufacturer in implementing Field Safety Corrective Actions (FSCA), e.g. fixing devices, recalling devices, and or withdrawing devices from the market. Such execution of FSCA may also be requested to the Distributor directly by a Competent Authority.
If the Distributor considers a medical device to present a serious risk to safety, they further are obliged to inform the Competent Authorities without delay. It should be noted here, that again procedures including definitions of serious risk are important to ensure proper implementation. Before informing Competent Authorities it is strongly recommended to include a liaison with the Manufacturer to ensure that their concerns are indeed correct.