
Co-authored with Preethi Philip
Under the MDD 93/42/EEC, the medical device industry lacked a serious level of transparency in the European Union as compared to the United States, where the FDA has all (510(k) & De Novo) clearances and (PMA) approvals publicly available including any incidents with medical devices (MAUDE). The scattered gathering of information in all European Member States was, and continues to be problematic in my personal view, and does not contribute to a safe and transparent industry.
Luckily, the MDR 2017/745 and the IVDR 2017/746 have introduced requirements for the development of a publicly accessible European Database on Medical Devices (EUDAMED). As we have seen, however, the development of EUDAMED is progressing slowly and its use is still not fully enforced.
In this article, we summarize the current status of EUDAMED and provide insight into the upcoming timelines and guidance on how to use EUDAMED.
5 Reasons Why We Need EUDAMED
Before discussing the timelines of EUDAMED, let’s assess why we should be happy with it and how it will benefit all parties in the medical devices field.
- Centralised Governance: As stressed above, there is a clear benefit from a regulator’s perspective to have a centralized system. It will grant regulator access to the full amount of information on devices available in the European Union Market, which previously, in earlier EUDAMED iterations (available to authorities only) was not;
- Regulatory and Safety Transparency: EUDAMED will support transparency amongst Economic Operators, for example, granting access to CE certificates from equivalent devices on the market, summaries of safety and clinical performance (for certain implantable Class IIb and Class III devices), which will allow industry to learn from each other’s safety risks and ensure that those are reviewed and embedded prior to conducting clinical investigations and prior to bringing devices onto the market. In my view, the latter should be true for classes of devices, and not be limited to implants and class III devices only;
- Device Classification Transparency: EUDAMED ‘finally’ will bring insight into device classification. This might not sound as a major step, however, the lack of transparency creates an unlevel playing field, where some devices, with identical intended purposes are marketed as Class IIa with one Notified Body and Class IIb with another. This inconsistency will hopefully be dealt with once and for all and create a level playing field;
- Transparency to End-Users: EUDAMED will provide transparency to the field of healthcare and patients, e.g. to investigate who has manufactured a device, which countries the devices are being made available to, what information is present in the summary of safety and clinical performance and whether a device is actually registered and CE marked;
- Replacement of Local Databases: Working in regulatory, I have had my share of experience needing to register in local member state databases (e.g. in Portugal, Spain, Italy, etc.), which are often available in local languages only, and which are all but straightforward, hopefully, EUDAMED will replace these databases once and for all.
EUDAMED’s Current Status
Even though the full use of EUDAMED isn’t enforced yet, some modules can already be used voluntarily. Moreover, several Competent Authorities have started to enforce the registration of Class I devices in EUDAMED as an alternative to registration with the local Competent Authority. As a manufacturer bringing to market a new Class I device, there is little reason (in my view) to avoid registration in EUDAMED straight away.
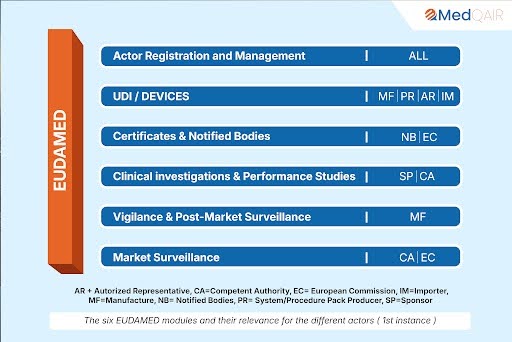
Overall EUDAMED is being rolled out through several modules. As of October 2024, the following modules have been made available:
- Actor Registration (Available since December 2020)
- UDI/Device Registration (Available since October 2021)
- Notified Bodies and Certificates (Available since October 2021, though some features like scrutiny mechanisms and the Clinical Evaluation Consultation Procedure (CECP) are still pending)
The remaining modules—covering Vigilance, Clinical Investigations & Performance Studies, and Market Surveillance—are currently still under development.
The EUDAMED system is set to be fully operational by 2027, with its complete mandatory use planned for manufacturers by 2029. Judging the current progression of the development of EUDAMED, and the continuous delays, we may expect the timeline to be pushed out further.
EUDAMED Timelines
The European Commission has outlined the following timeline for EUDAMED:
- May 2022: Actor Registration module became available
- Q2 2025 (Expected): Notification in the EU Official Journal that EUDAMED is functional
- Q4 2025 (Tentative): Mandatory use of EUDAMED for all stakeholders (regarding Actor Registration)
Please note that these dates are subject to change based on the European Commission’s assessment of the database’s full functionality.
How to Use EUDAMED
1. Access EUDAMED
- Visit the EUDAMED Portal and log in with your European Commission Authentication Service (ECAS) credentials. If you don’t have an ECAS account, you’ll need to create one.
- After logging in, you’ll be presented with different modules based on your role and the features available to you.
2. Register Your Organization
Before interacting with the system, ensure your organization is registered in EUDAMED:
- Go to the Actor Registration Module (ARM), where all economic operators (manufacturers, authorised representatives, importers, etc.) can register.
- Provide essential information, such as your company’s name, legal entity, and VAT/tax registration number.
- Once registered, you will receive a Single Registration Number (SRN), which requires an action (verification and approval) from your local Competent Authority. Once approval is granted a SRN is provided which you will need for further interactions with EUDAMED and regulatory authorities.
3. Submit Medical Device Information
After registering, manufacturers and other economic operators can submit details of the medical devices they are placing on the market:
- Navigate to the UDI/Device Registration Module. Here, you will enter the Unique Device Identification (UDI) for each device (Basic UDI & UDI-DI), along with relevant technical documentation.
- Input key data points such as device classification, intended use, risk category, and manufacturer information. This data must be updated regularly to ensure compliance.
Important Note:
With some registrations we noted that EUDAMED automatically assigns Annex X as the route for Conformity Assessment, requiring the upload of a CE certificate. Make sure to change it to Annex IX if that is your applied Conformity Assessment route, otherwise your product registration process will be blocked. This may have been resolved in the meantime.
4. Notified Body Interaction
A Notified Body (NB) is an organization appointed by an EU Member State (or other countries with specific agreements) to evaluate whether certain products meet regulatory standards before they are allowed on the market. If your medical device requires a conformity assessment by a NB, this step will apply*:
- The Notified Body (NB) responsible for your product will access EUDAMED’s Notified Bodies and Certificates Module to upload CE certificates and reports associated with the conformity assessment.
*= This is an independent step from device registration
5. Vigilance Reporting (not there yet)
Once your device is on the market, post-market surveillance and vigilance are critical aspects of compliance:
- Use the Vigilance and Post-market Surveillance Module to report serious incidents, safety concerns, and Field Safety Corrective Actions (FSCAs).
- Input detailed descriptions of the incident, the device involved, and actions taken. Notified Bodies and Competent Authorities will review these reports, and any necessary actions will be communicated through the system.
6. Clinical Investigation/Performance Study (not there yet)
For devices requiring clinical investigations (under MDR) or performance studies (under IVDR):
- Submit applications for clinical trials or studies via the Clinical Investigation and Performance Study Module. This is where the details of the planned study, including objectives, methodologies, and timelines, are uploaded for approval.
- Use this module to monitor the progress of the approval process and post results as required by the regulations.
7. Access Public Data
If you only need to search for information within EUDAMED, a specific EUDAMED login is not required. The public sections of EUDAMED are accessible without needing an account, allowing you to view important details. For users who are not actively involved in submitting data (such as manufacturers or notified bodies), the public section of EUDAMED offers valuable information on medical devices and certificates without needing registration or login credentials.
EUDAMED provides access to certain information for public users. Even without a login, anyone can access publicly available data:
- Navigate to the Public Portal to view registered devices, manufacturer data, and notified body certificates.
- This helps increase transparency, as healthcare professionals and the public can access essential data on medical devices being used across the EU.
8. Stay Compliant with Updates
It’s important to regularly update your device information in EUDAMED:
- Any changes in your device’s specifications, risk classification, or manufacturing process must be reflected in the database.
- Similarly, post-market surveillance reports, periodic safety updates, and changes in economic operator details must be continuously managed to stay compliant with MDR/IVDR.


