

On May 26, 2021, the Medical Device Regulation (MDR) 2017/745 went into full effect. One of the key changes included the introduction of enhanced traceability requirements for medical devices from manufacturers to final consumers across the whole supply chain. Part of the strategy from the European Commission to support traceability is the launch of the European Database on Medical Devices (EUDAMED), which is an IT system implemented to help achieve this goal. EUDAMED facilitates streamlining the enforcement of these rules, which is intended to improve the safety and transparency in the medical device industry.
UDI
Every medical device needs a Unique Device Identifier (UDI) so that it can be traced from production to the market. An authorized entity, such as GS1 or the Health Industry Business Communications Council (HIBCC), is required to issue this identification.
The UDI has three parts:
- Basic UDI-DI: The primary identifier for a group or family of products with the same intended purpose, risk level, and basic design and manufacturing characteristics.
- UDI-DI (Device Identifier): Unique to each device and/or packaging configuration and includes details about the device model, manufacturer, authorized representative, and other pertinent information.
- UDI-PI (Production Identifier): Records the various aspects of a device’s production, such as the lot number, serial number, manufacturing date, and expiration date.
EUDAMED Modules
EUDAMED is planned to include six modules, each with a specific purpose (note that not all modules are final yet). Manufacturers must register their devices and themselves with EUDAMED. Today, only economic operators – that is, Manufacturers, Importers, System/Procedure Pack Producers, and Authorized Representatives – can register to receive a Single Registration Number (SRN). Manufacturers need this SRN to enable them to register their devices with the relevant Competent Authority (CA) and start the conformity assessment process with a Notified Body (NB).
The six modules are:
- Actor registration
- UDI/Devices registration
- Notified Bodies and Certificates
- Clinical Investigations and performance studies
- Vigilance and post-market surveillance
- Market Surveillance
Which Countries are Registered in EUDAMED?
Operating under the EU’s Medical Device Regulation (MDR) and In-vitro Diagnostic Regulation (IVDR), EUDAMED serves multiple member states within the EU. Apart from these, the national competent authorities from a few other regions are also registered in the EUDAMED. Let’s take a look at all the countries that are registered in EUDAMED:
The 27 EU countries – The EU27 is made up of 27 countries, including Austria, Belgium, Bulgaria, Croatia, Cyprus, Czechia, Denmark, Estonia, Finland, France, Germany, Greece, Hungary, Ireland, Italy, Latvia, Lithuania, Luxembourg, Malta, Netherlands, Poland, Portugal, Romania, Slovakia, Slovenia, Spain, and Sweden
EFTA Countries – European Free Trade Association (EFTA) nations that are part of the European Economic Area (EEA) adhere to the EU MDR and are registered in EUDAMED. This covers Liechtenstein, Iceland, and Norway. Note that while Switzerland is part of EFTA, since the EU and Switzerland no longer have a mutual recognition agreement for medical devices as of May 26, 2021, the Swiss national competent authorities are not registered in EUDAMED.
Turkey: As a member of the Customs Union and a prospective EU member, Turkey largely conforms its medical device regulations to EU standards.
Northern Ireland: Following Brexit, Northern Ireland complies with EU medical device regulations under the Northern Ireland Protocol. This means that any medical devices that are put on the market in Northern Ireland have to comply with EU regulations, including registration in EUDAMED.
Now that we’ve reviewed the basics of EUDAMED and the countries that are registered in the database, let’s compare it to a few similar regulatory databases from different countries.
US - GUDID and Other FDA Medical Device Databases
Before talking about the FDA Medical Device Databases, we should look into the FDA Establishment Registration Database. The establishment registration database is used to register establishments that manufacture, prepare, and distribute medical devices in the United States. This registration is a prerequisite for listing medical devices with the FDA.
Once an establishment is registered, it is required to list all medical devices that it manufactures or processes. This includes providing details about each device, such as its intended use, and any applicable premarket submission numbers (510(k), De Novo, PMA, PDP, HDE).
Similarities between EUDAMED and GUDID:
Purpose – Both databases were created with the goal of improving the identification and traceability of medical devices, ensuring safety and transparency.
Manufacturer Requirements – Both mandate that manufacturers store critical data about their medical equipment in the respective databases.
UDI Structure – Both regions use a similar structure for UDI, and include the UDI-DI (Device Identifier), which serves as a link to both EUDAMED and GUDID. However, keep in mind that not all UDI issuing organisations may meet the requirements for both databases. Some suppliers may only provide UDIs that are compliant with one system and not the other. GS1, HIBCC, and ICCBA are recognized as issuing entities in every country except China.
Differences between EUDAMED and GUDID
- MAUDE – The MAUDE database focuses on postmarket surveillance in the US, collecting and reviewing reports of adverse incidents, malfunctions, and problems with medical device products.
- 510(k) – The 510(k) Database contains all premarket notifications (510(k) submissions). Medical device manufacturers are required to submit a 510(k) premarket submission to the FDA if they are launching a new product or have substantially modified an existing device to make a new one.
- Premarket Approvals (PMA) – Premarket approval (PMA) is the FDA’s scientific and regulatory evaluation process for assessing the efficacy and safety of Class III medical devices. Premarket approvals of high-risk Class III medical devices are listed in the database.
- De Novo – De novo offers a possible approach to classifying novel devices of low- to moderate-risk. The database contains De novo categorization orders.
- AccessGUDID (Global Unique Device Identification Database) – Contains essential identification details about medical devices that have unique device identifiers (UDI)
Switzerland - Swissdamed
Similarities between Swissdamed and EUDAMED
Regulatory Objectives – Both Swissdamed and EUDAMED aim to enhance the safety, traceability, and transparency of medical devices in their individual markets.
UDI – Both systems utilize UDI to improve medical device traceability, which facilitates product management and tracking.
Manufacturer Requirements – Both mandate that manufacturers keep thorough records and provide accurate information on their devices.
Differences between Swissdamed and EUDAMED
Modules – While EUDAMED has 6 modules, Swissdamed has 2 modules. They are:
- Actor Registration Module: This module allows Swiss manufacturers, authorized representatives, importers, and other business entities to register and manage their information.
- Device Registration Module: Manufacturers can use this module to register their medical devices before putting them on the market. It also involves registering the UDIs linked to the devices.
Geographical Scope
- EUDAMED: Operates within the EU and aligns with the EU Medical Device Regulation (MDR) and In Vitro Diagnostic Regulation (IVDR).
- Swissdamed: The Swiss equivalent of EUDAMED, created to align with the Swiss Medical Devices Ordinance (MedDO) and In Vitro Diagnostic Medical Devices Ordinance (IvDO).
UK - UK MDR
Similarities between EUDAMED and UK MDR
Device Classification – Both systems use risk-based classifications for medical devices, with categories that determine the regulatory pathway (UK MDR: Class I, IIa, IIb, III; EUDAMED: Class I, IIa, IIb, III).
UDI – A UDI system is incorporated into both frameworks to improve medical device identification and traceability.
Differences between EUDAMED and UK MDR
Regulatory Framework
- UK MDR: Following Brexit, UK MDR operates under the UK’s own regulatory framework, which is mostly in line with previous EU laws but has been modified for the UK market.
- EUDAMED: Operates in accordance with the EU Medical Device Regulation (MDR) and In Vitro Diagnostic Regulation (IVDR).
Database
- Under UK MDR, all medical device manufacturers are required to register in the Public Access Registration Database (PARD).
- EU MDR requires medical device manufacturers and certified devices to be registered in EUDAMED.
Vigilance Reporting
- When it comes to vigilance reporting concerning medical devices and in vitro diagnostics (IVDs), the MHRA relies largely on the Manufacturer’s On-Line Reporting Environment (MORE). MORE streamlines the submission of vigilance reports, including Manufacturer Incident Reports (MIRs), Field Safety Corrective Actions (FSCAs), and other safety-related documentation for medical devices.
- Article 92 of EU MDR mandates that all vigilance reports be submitted to EUDAMED.
Both EUDAMED and UK MDR are intended to efficiently regulate medical devices; but, owing to the UK’s exit from the EU, they operate under different frameworks. Even though they have comparable objectives and certain structural similarities, their regulatory procedures differ significantly.
China - National Medical Products Administration (NMPA)
Similarities between EUDAMED and NMPA
Device Registration – Both EUDAMED and NMPA require manufacturers to register their medical devices before they can be marketed. This includes providing detailed information about the devices, such as specifications and safety data.
UDI – Both EUDAMED and the NMPA have implemented UDI systems to improve traceability and safety. Devices must be labelled with a UDI that is recorded in the respective databases.
Post-Market Surveillance – Both databases facilitate post-market surveillance by collecting data on adverse events and device performance, helping to monitor device safety after they have been approved for use.
Differences Between EUDAMED and NMPA
Purpose
- EUDAMED facilitates the exchange of information between the European Commission and national competent authorities.
- NMPA regulates the safety, efficacy, and quality of medical devices within China.
Scope
- EUDAMED operates across multiple countries within the EU, aiming to improve coordination between the different Member States.
- NMPA largely focuses on China but is gradually aligning with international standards.
Database Structure – EUDAMED is composed of six interconnected modules, whereas NMPA is primarily a registration database with vigilance and UDI management.
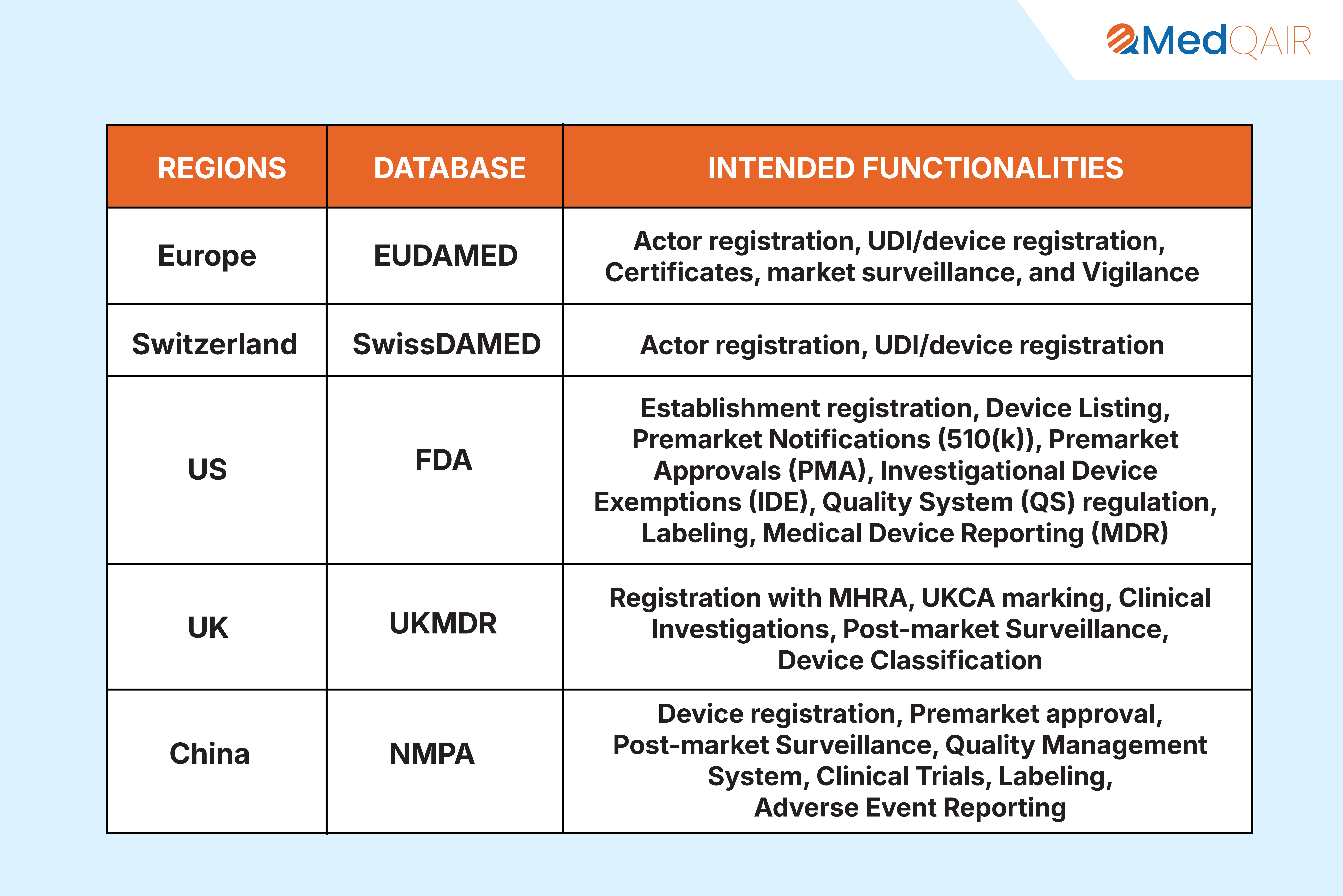
Here’s a table that compares each database and its intended functionalities.
Key Takeaways
In conclusion, EUDAMED is vital to improving the safety and transparency of medical devices in the EU and its member states. Comparing EUDAMED with other international regulatory databases allows us to find valuable synergies that promote better compliance and safety standards around the globe.
For manufacturers navigating this complex environment, partnering with an experienced and reliable authorised representative service provider can provide invaluable support. Our experts at MedQAIR can assist you with EUDAMED registration and regulatory compliance, enabling you to meet all regulatory requirements quickly and efficiently.
Get in touch with us to let us know how we can support your regulatory requirements!




